

治療目的/所見クラスⅠ クラスⅡa クラスⅡb
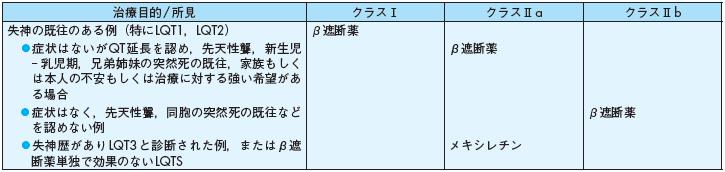
失神の既往のある例(特にLQT1,LQT2) β遮断薬
◦症状はないがQT 延長を認め,先天性聾,新生児︲ 乳児期,兄弟姉妹の突然死の既往,家族もしく
は本人の不安もしくは治療に対する強い希望がある場合
β遮断薬
◦症状はなく,先天性聾,同胞の突然死の既往などを認めない例
β遮断薬
◦失神歴がありLQT3と診断された例,またはβ遮断薬単独で効果のないLQTS
メキシレチン
失神の既往のある例(特にLQT1,LQT2) β遮断薬
◦症状はないがQT 延長を認め,先天性聾,新生児︲ 乳児期,兄弟姉妹の突然死の既往,家族もしく
は本人の不安もしくは治療に対する強い希望がある場合
β遮断薬
◦症状はなく,先天性聾,同胞の突然死の既往などを認めない例
β遮断薬
◦失神歴がありLQT3と診断された例,またはβ遮断薬単独で効果のないLQTS
メキシレチン
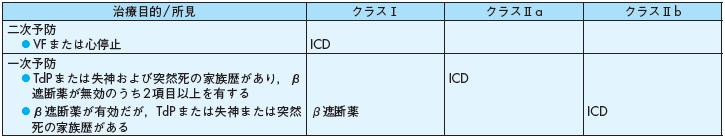
治療目的/所見クラスⅠ クラスⅡa クラスⅡb
二次予防
◦VF または心停止ICD
一次予防
◦ TdPまたは失神および突然死の家族歴があり,β遮断薬が無効のうち2項目以上を有する
ICD
◦β遮断薬が有効だが,TdPまたは失神または突然死の家族歴がある
β遮断薬ICD
二次予防
◦VF または心停止ICD
一次予防
◦ TdPまたは失神および突然死の家族歴があり,β遮断薬が無効のうち2項目以上を有する
ICD
◦β遮断薬が有効だが,TdPまたは失神または突然死の家族歴がある
β遮断薬ICD
11 先天性QT 延長症候群(LQTS)
Romano-Ward症候群とJervell and Lange-Nielsen症候群に代表される,QT間隔の延長とtorsade de pointes(TdP)という特徴的な心室性不整脈による
失神や突然死を来たす遺伝性不整脈疾患である435)-438).前者の頻度は1人/1000人,後者は1人/100万人とされる439).欧米では,年間8,000人の小児突
然死例のうち半数以上がLQTSによると想定されている.本症の10%は心停止が初発症状であり,予知と予防が極めて重要である438).運動,精神的興
奮,緊張,驚愕などで失神を来たした例では本疾患を疑うことが重要で,心電図,家族内の突然死や失神の有無などを参考に診断する439).
LQTSの原因は心筋のイオンチャネルや調節蛋白の機能異常により,現在までに12の関連遺伝子の異常が報告されている(LQT1からLQT12と呼ばれ
る).このうち,LQT1,2,3の順に多く,この3型で大部分が占められる192),193).臨床像の解析や治療もこれらの3 型で最も進んでおり,臨床像や
心電図所見は,3つの遺伝子異常によって異なる194)-197).
不整脈事故は男性では思春期以前に発生することが多く,女性より発生頻度が高い.思春期以降では女性患者の不整脈事故が多くなる440).小児期の
不整脈事故は,LQT1では男性のほうが女性よりもが多いが,LQT2およびLQT3で性差はない192).LQT1(112人),LQT2(72人),LQT3(62人)の生下
時から40歳までの心イベント(失神,心停止,突然死)の発生率は,それぞれ63%,46%,18%であるが,致死率は,LQT1とLQT2では4%であるのに対し,
LQT3では20%と高い192).
TdPの発症リスクは,LQT2ではQTc > 500msec以上,LQT3では男性で高い192).またβ遮断薬治療にもかかわらず失神を繰り返す例や388),441),
突然死の家族歴がある場合は高危険群となる196).TdPの発症は,LQT1では運動,特に水泳中,LQT2では精神ストレス,突然の聴覚刺激や出産直後に,
LQT3では睡眠中に多い193),442).LQT2では孔(pore)領域の遺伝子異常を有する群の方が,その他の領域に遺伝子異常を有する群よりTdPが発症する危
険は高く186),LQT1では交感神経活動の影響を受けやすくなる199).またLQT1では膜ドメインに異常が認められる方が,C末やN末に遺伝子異常を有するも
のに比べ予後が悪いことが報告されている443).
LQTSの遺伝子異常が確認された発端者の家族には,症状やQT延長を認めないにもかかわらず33%に発端者と同様の遺伝子異常が認められる188).
これはcarrierの存在を示す以外に,Kチャネルを抑制する薬剤の使用時や徐脈時にTdPを来たす潜在的な危険群の存在を示している.ICDの適応は突然死
の家族歴,TdPの発生や失神発作の既往,β遮断薬の有効性などを参考に決定される429)(表11,12).
表11 先天性QT 延長症候群(LQTS)における突然死予防
表12 先天性QT 延長症候群(LQTS)の薬物適応
注)運動とQT延長作用を有する薬物(抗不整脈薬,三環系抗うつ薬,抗ヒスタミン薬など)は全例で禁忌.
心臓突然死の予知と予防法のガイドライン(2010年改訂版)
Guidelines for Risks and Prevention of Sudden Cardiac Death(JCS 2010)
Guidelines for Risks and Prevention of Sudden Cardiac Death(JCS 2010)